


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Amplification of Multiple Receptor Tyrosine Kinase Pathways in a Patient with Metastatic Castration-Resistant Prostate Cancer with Disseminated Intravascular Coagulation (DIC)
Tingrui Wang1,Zonghui Ding2,Buqing Ni3,Jeffrey Wang4,Jue Wang5
2.Department of Biochemistry and Molecular Biology, Mayo Clinic Arizona, Scottsdale, AZ, USA.
3.Baylor University, Waco, TX, USA.
4.University of Arizona Cancer Center at Dignity Health St. Joseph's, Phoenix, AZ, USA.
Copyright : © 2017 . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Background: Disseminated intravascular coagulation (DIC) has long been associated with prostate cancer and prognosis is very poor. The molecular profiling of DIC with prostate cancer has not been reported to date. Understanding the molecular mechanisms of castration resistant prostate cancer (CRPC) and associated DIC may improve response to therapy and clinical trial designs.
Methods:
A 72-year-old man with past medical history of metastatic castration resistant prostate cancer developed progressive disease with DIC. Patient was treated with mitoxantrone, flutamide and ketoconazole. The next day, PSA and D-dimer was reduced from 4697 ng/ml to 1842 ng/ml and 27, 645 to 7871 ng/ml, respectively. Two months later, patient presented in emergency department with altered mental -status. His head CT scan showed large mixture of intraparenchymal and subarachnoid hemorrhage.
Results:
Guardant 360 biopsy-free TM tumor sequencing showed AR mutation, AR, MET, EGFR, FGFR1 amplification.
Conclusions: An in-depth analysis of tumor molecular profile may facilitate drug development and provide new insights into CRPC and its related DIC.
1. Introduction
Disseminated intravascular coagulation (DIC) is characterized by activation of coagulation which results in thrombosis organ failure and severe bleeding. DIC may be caused by infection, sepsis, solid cancers, leukemia, and obstetric diseases. The incidence rate of DIC is 13–30% in metastatic prostate cancer [1, 2]. Patients with prostate cancer has been observed to develop a life threatening DIC and may develop systemic and intracranial bleeding due to excessive fibrinolysis. Current understanding on mechanisms of DIC in prostate cancer is limited and the knowledge linking prostate cancer progression and DIC is lacking.
Prostate cancer is the most commonly diagnosed male cancer and is the third leading cause of cancer-related deaths in men. Approximately 161,360 American men will be diagnosed with prostate cancer this year, and more than 26,000 can expect to die from their disease[3]. Androgen deprivation therapy (ADT) is considered first-line treatment, which provides initial benefits, however, progression to castration-resistant prostate cancer (CRPC) invertible disease despite medical or surgical castration. Understanding mechanism of resistance is essential to develop future treatment. [4] The molecular profiling of DIC with prostate cancer has not been reported to date, further investigation may provide insights on the association of the development of DIC and progression of CRPC.
In this study, we report molecular findings from next generation sequencing (NGS) of circulating DNA in a prostate cancer patient with DIC. Our findings of gene amplification both AR and multiple oncogenic receptor tyrosine kinases (including c-Met and EGFR) may shed some light on the association of prostate cancer and DIC. In addition, we reviewed the clinical presentation and management of 81 prostate cancer patients with DIC.
2. Results
A 72-year-old man had a known medical history of metastatic castration resistant prostate cancer with progressive disease after multiple lines of therapies including biclutamide, lupron,Provenge immunotherapy, enzalutamide, abiraterone, docetaxel. In September 2016, patient presented with fatigue and generalized weakness. Patient Physical exam was unremarkable except for multiple skin brushes and petechiae. Laboratory results showed hemoglobin (Hb) 6.8 gm/dl, hematocrit 20.1% (normal range: 40-52%) platelets 39 x109/L. Prothrombin time (PT) seconds was 17.5 s (normal range: 10–12.3), activated partial thromboplastin time (aPTT) 36.3s (normal range: 25.5–36), fi 36.3s ( 325 mg/dl, and D-dimer 27,645 ng/ml (normal range: 150-243). PSA was 4697 ng/ml (normal range: 0.05-4). CT head showed no acute intracranial hemorrhage and no other acute intracranial abnormality.
Patient was treated with blood transfusion, mitoxantrone 12 mg/m2 intravenously monthly and flutamide 250 po mg q8hr and ketoconazole po 200mg q8hr. The next day, PSA and D-dimer was reduced from 4697 ng/ml to 1842 ng/ml and 27,645 to 7871 ng/ml respectively. Guardant 360 biopsy-free TM tumor sequencing showed multiple genomic alterations including androgen receptor (AR) mutation, AR amplification, EGFR, FGFR1 and MET amplification.
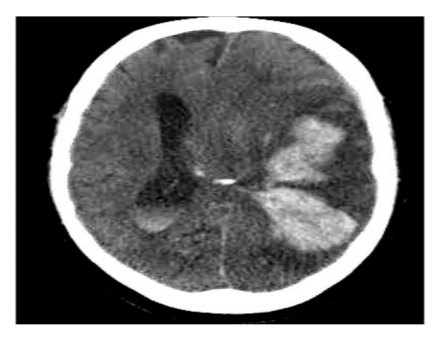
Two months later, patient was admitted to hospital for altered mental status. On assessment in the emergency department, his head CT scan showed large mixture of intraparenchymal and subarachnoid hemorrhage involving large portion of the left cerebral hemisphere, where associated severe mass effect upon the left lateral ventricle and left temporal lobe with prominent approximate 1.5 cm midline shift to the right (Figure 1). Laboratory results showed hemoglobin (Hb) 6.8 gm/dl, hematocrit 20.3% (40-52%), platelets 18 x109/L. PT was 22.2 s (10–12.3), activated PTT 31.9 s (25.5–36), fibrinogen 235 mg/dl, D-dimer 7463 ng/ml (150-243). The patient was intubated, admitted to the ICU.
Figure1. Head CT scan showed large mixture of intraparenchymal and subarachnoid hemorrhage involving large portion of the left cerebral hemisphere. Associated severe mass effect upon the left lateral ventricle and left temporal lobe with prominent approximate 1.5 cm midline shift to the right
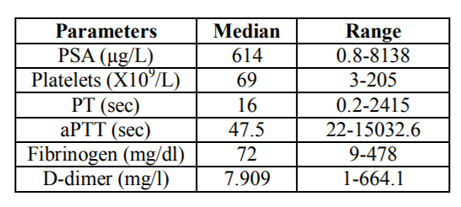
Through the literature review by searching the database for previous published journals, we were able to additionally reviewed the reported clinical features of 81 prostate cancer patients with DIC (age range: 43-92 years, median age: 68 years, Table 1). 79 out of 81 patients were known to have stage IV prostate cancer. Ninety-one percent of the patients (74/81) had metastases when they developed DIC. The level of PSA at the diagnosis of DIC was at average of 614 µg/L. The median value of platelet counts was 69 x103/ µL. The median survival time of these patients was 4.5 months. Patients had elevated D-dimers, PT, aPTT and decreased fibrinogen and platelet (Table 2).
Table1
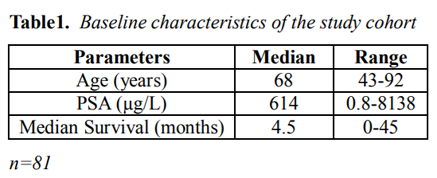
Table2 Laboratory parameters of prostate cancer patients with DIC
Bleeding induced by DIC included subcutaneous bleeding in 52 patients (64%), hematuria in 22 (27%), epistaxisin 21 (26%), invasive procedure (incision, biopsy, trauma, operation) bleeding in 20 (25%), gastrointestinal bleeding in 18 (22%), oral in 13 (16%), and cerebral bleeding in 6 (7%) (Table3)
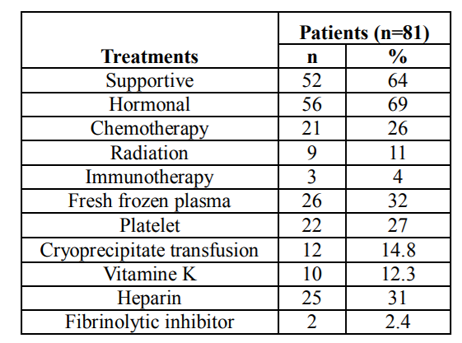
Management of DIC varies widely within this study. 69% (56/81) patients received anti androgen hormonal therapy, 64% (52/81) supportive care, 32.1% (26/81) fresh frozen plasma, 27.1% (22/81) platelet, 14.8% (12/81) cryoprecipitate transfusion, 12.3% (10/81) vitamin K, 31% (25/81) heparin use, 26% chemotherapy(21/81), 11% radiation therapy (9/81), 4% immunotherapy (3/81), and 2.4% (2/81) fibrinolytic inhibitor (aminocaproic acid). (Table 4)
Table3 Sites of bleeding induced by DIC
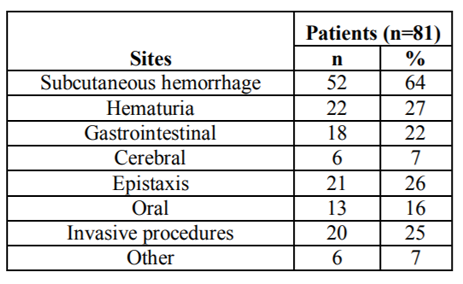
Table4 Types of treatments for prostate cancer related DIC
3. Discussion
Prostate cancer is the most commonly diagnosed cancer in men. Androgen deprivation therapy is the standard treatment for locally advanced or metastatic prostatic cancer which provides initial benefits. Majority of patients will progress to castration resistant prostate cancer within three years. Disseminated intravascular coagulation has long been associated with prostate cancer development and prognosis is very poor. A retrospective analysis of prostate cancer revealed that DIC was mostly seen in patients with high-grade prostate cancer and 93% patients were castration resistant [5].
Androgen axis plays an important role in the CRPC development. Five theories proposed on the causes of development of aggressive castration resistant cancer: hypersensitive,outlaw, promiscuous, coactivators and corepressors, bypass pathway[4]. Thirty to eighty percent of CRPC cell lines show amplification of the AR and twenty percent of CRPC metastases have evidence of AR amplification [6, 7]. AR Splice variants of AR (AR-Vs), which lacks the ligand binding domain (LBD) and is constitutively active [8-11]. Various AR point mutations have been identified that lead to increased AR activity in the presence of low androgen level which has been associated with more aggressive disease [12, 13]. AR recruits co-activators or co-repressors to enhance or repress transcriptional activity. Co- regulators identified as molecular chaperones modulate the protein through phosphorylation, acetylation, methylation. Growth factor receptors such as EGFR control survival pathways, enhance AR activity and promote androgen independence [4].
Genomic profiling is a useful tool for understanding the molecular mechanism and etiology of CRPC and cancer related DIC. ctDNA analysis provides a way to identify mutational mechanisms of drug resistance in CRPC. In our case, patient ctDNA demonstrates AR mutation, AR amplification, EGFR, FGFR1 amplification which explains his resistance to therapy. These newly identified mutations might offer novel target treatment that can overcome resistance.
DIC is a devastating complication of CRPC leading to excessive fibrinolysis and bleeding. It is characterized by hypofibrinogenemia with decreased platelet, increased D-dimer and prolonged PT/aPTT. If untreated, the hemorrhage risk is high and survival time is very short. The most important principal to manage prostate cancer with DIC is to treat underlying disease. Improvement in DIC has been shown in active cancer therapies such as androgen suppressive therapies. [4-16, 18-21] and chemothepy [17] Supportive blood products aimed at reversing hemostatic derangements and reducing bleeding. In general, platelet transfusion is administered in DIC patients with active bleeding and a platelet count less than 50 x109/L. Fresh frozen plasma and cryoprecipitate replete clotting factor and fibrinogen respectively. Fibrinogen deficiency (less than 1g/dl) can be corrected with the administration of purified fibrinogen concentrates or cryoprecipitate. Fibrinolytic inhibitor aminocaproic acid is effective in treating bleeding. However, cases complicated with severe thrombosis have been documented [22]. Patients with DIC are at high risk of venous thromboembolism (VTE). The administration of heparin is beneficial in non-symptomatic DIC to prevent deep vein thrombosis (DVT), however should be avoided in active bleeding[23].
The mechanisms underlying the association between CRPC and DIC are multifactorial and under investigation. Expression of procoagulant proteins or cancer procoagulant may provoke thrombin generation which results in fibrinolysis and anti-fibrinolytic response. Tissue factor (TF) level is an important marker to predict the development of DIC in patients with cancer. A significant elevation of TF level was observed in DIC associated cancer, however no significant elevation in leukemia patients[24]. TF exposed to circulation is the culprit to initiate of ongoing DIC. The continuous exposure to TF exhausts the tissue factor pathway inhibitor (TFPI) which cause thrombin generation, and activation of factor XI and fibrin generation[25]. In glioblastoma multiforme (GBM), cancer cells overexpress tissue factor and interact with coagulation system which is driven by genetic transformation through expression of epidermal growth factor receptor (EGFR)[26]. Rak J. et al. demonstrated that upregulation of tissue factor is caused by oncogenic events including activation of K-ras and epidermal growth factor receptor (EGFR) [27]. The changes on tissue factor expression may cause a systemic hypercoagulable state and lead to tumor progression [28-31].
MET oncogene encodes a tyrosine kinase receptor for growth factor. MET and its ligand hepatocyte growth factor (HGF) play an important role in the development and progression of CRPC. Activation of the HGF/c-Met axis is a late event in prostate cancer progression. Prominent expression of c-Met was observed in advanced prostate cancer with bone metastases[32].
Recent investigation suggests activation of oncogenes RAS or MET might induce the expression of genes controlling hemostasis and cause thrombotic event. Fibrin formation results in an advantage for prostate cancer cells by providing scaffold to promote tumor invasion and growth [33]. Boccaccio et al. study shows MET oncogene was thrombogenic. All mice injected with MET oncogene developed thrombi and later developed DIC [31]. MET oncogene activation and amplification results in up regulation of cyclooxygenase(COX-2) and plasminogen activator inhibitor type 1(PAI-1). The elevation level of COX-2 and PAI-1 induced fibrinolysis and activation of platelets which results in DIC. Administration of inhibitors for PAI-1 and COX-2 caused decreased level of D-dimer[31]. Cabozantinib (XL184) is a predominantly MET tyrosine kinase inhibitor which demonstrates potent antitumor effects in CRPC. A variety of xenograft models demonstrated that cabozantinib inhibits MET signaling and induces apoptosis of cancer cells [34]. The study of phase II trial of cabozantinib showed resolution of bone metastasis in CRPC within six weeks[32].
4. Conclusion
DIC is a devastating complication of prostate cancer and prognosis is extremely poor. The fundamental management for this condition is to control underlying prostate cancer Our findings of amplification AR and multiple oncogenic receptor tyrosine kinases are very interesting, additional research are warranted. New treatment strategies for disseminated intravascular coagulation may potentially emerge based on understanding of the pathophysiology.
References
- Deme, D., et al., Metastatic prostate cancer complicated with chronic disseminated intravascular coagulopathy causing acute renal failure, mimicking thrombotic throm bocytopenic purpura and hemolytic uremic syndrome: pathomechanism, differential diagnosis and therapy related to a case. Magy Onkol, 2010. 54(4): p. 351-7.
- Smith, J.A., Jr., M.S. Soloway, and M.J. Young, Complications of advanced prostate cancer. Urology, 1999. 54(6A Suppl): p. 8-14.
- Siegel, R.L., K.D. Miller, and A. Jemal, Cancer Statistics, 2017. CA Cancer J Clin, 2017. 67(1): p. 7-30.
- Chandrasekar, T., et al., Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol, 2015. 4(3): p. 365-80.
- Hyman, D.M., G.A. Soff, and L.J. Kampel, Disseminated intravascular coagulation with excessive fibrinolysis in prostate cancer: a case series and review of the literature. Oncology, 2011. 81(2): p. 119-25.
- Liu, W., et al., Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia, 2008. 10(8): p. 897-907.
- Taylor, B.S., et al., Integrative genomic profiling of human prostate cancer. Cancer Cell, 2010. 18(1): p. 11-22.
- Dehm, S.M. and D.J. Tindall, Alternatively spliced androgen receptor variants. Endocr Relat Cancer, 2011. 18(5): p. R183-96.
- Guo, Z., et al., A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res, 2009. 69(6): p. 2305-13.
- Hu, R., et al., Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res, 2009. 69(1): p. 16-22.
- Sun, S., et al., Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest, 2010. 120(8): p. 2715-30.
- Makridakis, N.M., E. di Salle, and J.K. Reichardt, Biochemical and pharmacogenetic dissection of human steroid 5 alpha-reductase type II. Pharmacogenetics, 2000. 10(5): p. 407-13.
- Scariano, J.K., et al., The SRD5A2 V89L polymorphism is associated with severity of disease in men with early onset prostate cancer. Prostate, 2008. 68(16): p. 1798-805.
- Litt, M.R., W.R. Bell, and H.A. Lepor, Disseminated intravascular coagulation in prostatic carcinoma reversed by ketoconazole. JAMA, 1987. 258(10): p. 1361-2.
- Lowe, F.C. and W.J. Somers, The use of ketoconazole in the emergency management of disseminated intravascular coagulation due to metastatic prostatic cancer. J Urol, 1987. 137(5): p. 1000-2.
- Chakrapee-Sirisuk, S., et al., Ketoconazole and Flutamide in the treatment of disseminated intravascular clotting from prostate cancer: a case report and review. J Med Assoc Thai, 2001. 84(10): p. 1495-501.
- Smith, M.R., Successful treatment with mitoxantrone chemotherapy of acute disseminated intravascular coagulation due to metastatic androgen independent prostate cancer. J Urol, 2000. 163(1): p. 248.
- Doll, D.C. and J.W. Yarbro, Vascular toxicity associated with chemotherapy and hormonotherapy. Curr Opin Oncol, 1994. 6(4):p. 345-50.
- Al-Mondhiry, H., et al., Hemostatic effects of hormonal stimulation in patients with metastatic prostate cancer. Am J Hematol, 1988. 28(3): p. 141-5.
- Cornfield, D.B. and R.E. Rossman, Diethylstilbestrol-diphosphate-induced disseminated intravascular coagulation in prostatic carcinoma. South Med J, 1982. 75(2): p. 248-9.
- Bern, M.M., Coagulopathy, following medical therapy, for carcinoma of the prostate. Hematology, 2005. 10(1): p. 65-8.
- Brown, J.E., et al., All-trans retinoic acid (ATRA) and tranexamic acid: a potentially fatal combination in acute promyelocytic leukaemia. Br J Haematol, 2000. 110(4): p. 1010-2.
- Wada, H., T. Matsumoto, and Y. Yamashita, Diagnosis and treatment of disseminated intravascular coagulation (DIC) according to four DIC guidelines. J Intensive Care, 2014. 2(1): p. 15.
- Asakura, H., et al., Role of tissue factor in disseminated intravascular coagulation. Thromb Res, 1995. 80(3): p. 217-24.
- Osterud, B. and E. Bjorklid, The tissue factor pathway in disseminated intravascular coagulation. Semin Thromb Hemost, 2001. 27(6): p. 605-17.
- Magnus, N., D. Garnier, and J. Rak, Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood, 2010.116(5): p. 815-8.
- Rak, J., P. Klement, and J. Yu, Genetic determinants of cancer coagulopathy, angiogenesis and disease progression. Vnitr Lek, 2006. 52 Suppl 1: p. 135-8.
- Yu, J.L., et al., Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood, 2005. 105(4): p. 1734-41.
- Rong, Y., et al., PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res, 2005. 65(4): p. 1406-13.
- Tallman, M.S., et al., Effects of all-trans retinoic acid or chemotherapy on the molecular regulation of systemic blood coagulation and fibrinolysis in patients with acute promyelocytic leukemia. J Thromb Haemost, 2004. 2(8): p. 1341-50.
- Boccaccio, C., et al., The MET oncogene drives a genetic programme linking cancer to haemostasis. Nature, 2005. 434(7031): p. 396-400.
- Varkaris, A., et al., The role of HGF/c-Met signaling in prostate cancer progression and c-Met inhibitors in clinical trials. Expert Opin Investig Drugs, 2011. 20(12): p. 1677-84.
- Boccaccio, C. and P.M. Comoglio, Genetic link between cancer and thrombosis. J Clin Oncol, 2009. 27(29): p. 4827-33.
- Yakes, F.M., et al., Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther, 2011. 10(12): p. 2298-308.