


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
ARC Journal of Hematology
Volume-1 Issue-1, 2016, Page No: 1-5
Differentiation, Diagnosis, and Treatment of Atypical Hemolytic Uremic Syndrome: A Mini-Review
Mathew Justus ENS ¹,Samir Dalia2*;
1.Des Monies University College of Medicine, Des Monies, Iowa,USA
2.Mercy Clinic Oncology and Hematology, Joplin, MO,USA
2.Mercy Clinic Oncology and Hematology, Joplin, MO,USA
Citation : Dalia S, ENS MJ. Differentiation, Diagnosis, and Treatment of Atypical Hemolytic Uremic Syndrome: A Mini-Review. ARC Journal of Hematology. 2016;1(1):1–5.
Copyright : © 2016 Dalia S. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Atypical HUS is a thrombotic microangiopathy that can lead to morbidity and mortality if not diagnosed in a timely fashion. Like other thrombotic microangiopathies atypical HUS can have many different clinic symptoms including hemolytic anemia, thrombocytopenia and acute renal failure. With prompt diagnosis, interventions such as plasma exchange and/or eculizumab can be used in order to reverse disease progression. In this review we aim to try to help clinicians better understand the differences between the major thrombotic microangiopathies, learn how to better diagnosis atypical HUS and discuss the treatment of atypical HUS.
Keywords: Atypical HUS, microangiopathic hemolytic anemia, TMA, thrombocytopenia, renal failure, Eculizumab
1.Introduction
Thrombotic microangiopathy (TMA) is a term used to describe disorders involving microangiopathic hemolytic anemia (MHA), thrombocytopenia, and microvascular thrombosis leading to dysfunction of one or more organs. The classic forms of TMA include hemolytic uremic syndrome caused by Shiga-like toxin producing strains of E. coli (STEC-HUS) and thrombotic thrombocytopenic purpura (TTP), as well as disseminated intravascular coagulation (DIC). Atypical hemolytic uremic syndrome (aHUS) is another TMA. Long thought to be a variant of STEC-HUS or TTP, aHUS was recognized as a distinct entity is the 1970’s ,. In this article, the pathophysiology that differentiates aHUS from other forms of TMA, diagnosis of aHUS, and treatment of aHUS will be reviewed.
2.Differentiation
TMAs present with common clinical characteristics including hemolytic anemia and thrombocytopenia. LDH is usually elevated with a decreased haptoglobin. Since TTP, DIC, STEC-HUS, and aHUS can all present similarly, it is important to be able to differentiate these diseases in a timely manner to provide adequate treatment. While the presentations are similar, the pathophysiology is different, and there are subtle differences that can make clinical diagnosis simpler. We discuss the differences in diagnosis of the major TMAs below and table 1 summarizes the mechanism, presentation, and initial management of DIC, TTP, STEC-HUS, and aHUS.
3.Disseminated Intravascular Coagulation
DIC is a secondary TMA that presents in a variety of ways. As DIC is not a primary disease, DIC needs to be ruled out in any patient with a TMA. Conditions known to cause DIC include sepsis, trauma, malignancy, obstetric emergencies, and other less common causes such as AV malformations, transfusion reactions, burns, toxins, and amphetamine overdose. DIC presents, along with symptoms of the underlying condition, with bleeding in a majority, and rarely with renal, hepatic, and respiratory dysfunction. Lab work-up reveals increased PT/PTT, thrombocytopenia, elevated D-dimer, normal to low fibrinogen, and increased fibrin-split products. Treatment is correction of the underlying cause and supportive treatment. Packed red blood cells may be needed, as well as platelets if platelet counts drop below 10 x 109/L. Fresh frozen plasma can be used to treat coagulopathies in patients who are bleeding and cryoprecipitate is generally given to keep fibrinogen over 100mg/dL³
4.Thrombotic Thrombocytopenic Purpura
TTP is classically thought of as the pentad of thrombocytopenia, microangiopathic hemolytic anemia, neurologic symptoms, kidney failure, and fever, however this is true in less than 20% of cases [4]. For an initial clinical diagnosis, MHA and thrombocytopenia are required. Focal neurologic deficits are common, presenting as simple headache to stroke-like symptoms. Renal impairment such as mild proteinuria is common; however, renal failure is rare. Other more rare symptoms can occur secondary to cardiac, gastrointestinal, or retinal thrombi. Coppo determined that the more severe the ADAMTS-13 deficiency, the more severe the thrombocytopenia and the less severe the renal dysfunction, as determined by serum creatinine [5,6] . Treatment is started empirically with plasma exchange of 1-1.5 times plasma volume daily until platelet counts and hemolysis stops. Additionally, prednisone 1mg/kg/day orally or methylprednisolone 125mg intravenous twice daily in addition to plasma exchange has been shown to improve outcomes[7] . Early trials have shown rituximab, an anti-B-cell antibody, to be effective in combination with plasma exchange for refractory TTP [8]. Treatment of congenital TTP consists of recurrent infusions of fresh frozen plasma, which contains ADAMTS-13. Treatment for secondary TTP is treatment of the underlying cause along with plasma exchange.
Laboratory confirmation of TTP can be made by testing ADAMTS-13 levels. Available assays cannot accurately measure levels below 10% of normal, which is the cutoff for severe deficiency. Currently, there are only a few known disease states that lead to severe ADAMTS-13 deficiency—DIC, malignancy, and sepsis, along with TTP. Therefore, levels below the 10% threshold, in the absence of the previously mentioned disease states, have high positive predictive value for TTP. Unfortunately, however, the assay is not currently readily available for acute diagnosis, and is only useful after the fact. Once a diagnosis is confirmed, it may also be necessary to test for mutations leading to congenital TTP, as these patients will require ongoing treatment.
5.Hemolytic Uremic Syndromes
Both classic HUS and atypical HUS (aHUS) are defined by acute kidney injury, thrombocytopenia, and microangiopathic hemolysis. Classic HUS is caused by Shiga-like toxin producing strains of E. coli (STEC) in about 90% of cases, and thus has recently been referred to as STEC-HUS. Less commonly, HUS may be caused by Shigella dysenteriae type I. It most commonly occurs in children under the age of 10 years. STEC-HUS classically presents with bloody diarrhea 5-10 days prior to the onset of symptoms of renal dysfunction, thrombocytopenia, MHA, and variable levels of neurologic symptoms. It is hypothesized that neutrophils may transport Shiga-like toxins around the body [9]. Once out of the gut, Shiga-like toxins preferentially target the tubular epithelium, leading to cellular apoptosis, tubular necrosis, and thrombotic microangiopathy[10] . The TMA leads to kidney damage, as well as other symptoms, however, no thrombi have been found in the brains of patients suffering neurological symptoms, leading to many untested hypotheses [11]. Diagnosis is clinical, usually confirmed with positive serology for a causative organism. Treatment for STEC-HUS is supportive in nature, with no antibiotics indicated for STEC or S. dysenteriae type I. Some trials are underway in Germany exploring the use of eculizumab in STEC-HUS to prevent neurologic sequelae.
While presenting symptoms are similar, aHUS can be differentiated from STEC-HUS by the lack of preceding diarrheal illness. It is thought to occur at a rate of 3 per million in children under age 18, and at an even lower rate in adults[12]. aHUS has been determined to result from defects in the complement cascade. Three different types of molecular defects have been associated with aHUS: inactivation of complement regulators (CFH, MCP, CFI, or THBD), gain-of-function mutations of activators of complement (CFB or C3), and autoantibodies against CFH1. As most of these changes are genetic in nature, most cases present before the age of 10, however, some patients present later, even as late as 60 years of age[13]. The end result is uncontrolled complement activation, leading to formation of the membrane attack complex (MAC). The MAC in turn causes endothelial injury, leading to thrombosis. The combination of endothelial injury and thrombosis cause ischemia leading to organ injury, stenosis leading to increased shear stress and RBC fragmentation, and thrombocytopenia. The stenosis results in potentially severe hypertension. Renal failure, secondary to thrombi and ischemia, is severe in aHUS, often requiring dialysis differentiating it from the mild renal failure that can be seen in TTP. Increases in vascular permeability occur in about one third of patients with aHUS, leading to cerebral and pulmonary edema, pleural and pericardial effusion, and ascites and anasarca, complications which are relatively rare in TTP[1].
6.Diagnosis of AHUS
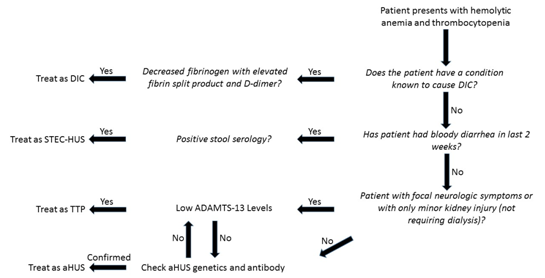
Clinically, symptoms of DIC, TTP, STEC-HUS, and aHUS can appear very similar and symptoms can overlap. However, there are some generalizations that can help to make a diagnosis, and lab tests are becoming available that can confirm diagnosis. The flow sheet below explains a path to diagnosis.
As mentioned previously, DIC can be ruled out in the absence of an underlying condition known to cause DIC with normal coagulation tests and normal fibrinogen. In the absence of diarrhea, STEC-HUS is doubtful, and a negative stool serology can help to it rule out. The more difficult pair to delineate is TTP versus aHUS. Comparatively, TTP is more likely to cause focal neurological defects and have lower platelet counts, with a lower tendency for acute kidney failure. aHUS is more likely to cause acute kidney failure requiring dialysis, hypertension, and conditions associated with increased vascular permeability.
Symptoms often overlap, and there are no pathognomonic symptoms for either condition. As differentiating aHUS and TTP clinically can be difficult, lab testing can be useful. As mentioned previously, ADAMTS-13 levels are an easy way to diagnose TTP, but often take too long to return to be useful in the acute setting. In the absence of ADAMTS-13 labs, tests are less conclusive. Coppo et al determined that patients with severe ADAMTS-13 deficiency are more likely to have a platelet count less than 30 x 109/L and serum creatinine less than 2.26 mg/L, with a 98% positive predictive value if both values are met, and an 85% positive predictive value if one is met, in the setting of a TMA [5,6]. If platelets are above 30 x 109/L, and creatinine tops 2.26, ADAMTS-13 should be sent for more guidance. If levels return normal, the presumptive diagnosis should become aHUS.
After TTP is no longer the presumptive diagnosis, direct tests for aHUS can be done. Currently, testing is available for antibodies to CFH, as well as genetic testing for complement dysfunction, however, not all patients with diagnosed aHUS have identifiable antibodies or genetic mutations. At this point, aHUS often remains a diagnosis of exclusion after DIC, STEC-HUS, and TTP have all be effectively ruled out.
Figure 1 .Pathway to diagnosis of different TMAs
7.Treatment Of AHUS
Since aHUS presents in a similar fashion as TTP, initial treatment is usually plasma exchange with or without corticosteroids. In the past plasma exchange was standard of care, however, results were poor. Results have been reported of 40% of children with aHUS, and 66% of adults with aHUS reaching end-stage renal disease by 5 years after initial diagnosis, and up to 50% of adults reach end-stage renal failure on initial presentation . Renal transplant, secondary to renal failure, was often unsuccessful in aHUS patients, due to recurrence of the disease, since the underlying defect was not corrected. It was later found that dual liver/kidney transplant had better outcomes due to correcting the complement dysfunction.
Several treatments have been tested, but not thoroughly studied. Rituximab has been used for cases of aHUS involving CFH antibodies to allow for transplants, by Kwon in 2008 and Le Quintrec in 2009 [15,16] . In 2010, Boyer described 3 pediatric patients treated for CFH antibodies with pulsed cyclophosphamide in addition to plasma exchange and corticosteroids, who all achieved clinical remission .
Eculizumab, a humanized monoclonal antibody of complement C5 that was first studied in and approved for paroxysmal nocturnal hemoglobinuria, a disease also caused by inappropriate complement activation is also a treatment for aHUS with the potential to improve renal function. It was first used to treat aHUS in 2009, and approved by the FDA in 2011. By binding to and preventing cleavage ofC5, eculizumab prevents downstream components of complement from being activated, thus preventing MAC formation. In adults, eculizumab is given as a 900 mg IV infusion weekly for 4 weeks, a 1200 mg dose on week 5, and then transitioned to 1200 mg every 2 weeks. In children, the dose in altered for body weight, with a similar induction and maintenance schedule. Patients who have not been vaccinated for N. meningitidis should be vaccinated 14 days prior to initiating treatment, or should be treated with antibiotics for 14 days after receiving the vaccination, due to reliance on complement to fight off encapsulated bacteria.
Prospective studies in adults with chronic aHUS have shown patients receiving eculizumab maintain normal platelet levels, improve glomerular filtration rate, and were sometimes able to stop dialysis . Two trials, one on patients with worsening aHUS (n=17) and one on patients with chronic aHUS (n=20), conducted by Licht et al over 2 years, showed that that 88% and 90% of patients had normal platelet counts at 2 years, 88% and 95% were TMA event free at 2 years, and 76% and 55% had a decrease of in serum creatinine greater than or equal to 25% of baseline at 2 years . A third retrospective pediatric trial (n=19) by the same group established the safety and effectiveness in children. Treatment with eculizumab has not been compared directly with plasma exchange.
As of the date of this article, treatment is considered to be lifelong. Currently, studies about discontinuation are underway; with case reports by Ardissino demonstrating discontinuation of therapy may be possible without greatly increased risk to patients 20. 10 patients who had achieved remission based off a battery of laboratory and clinical criteria were allowed to discontinue treatment with eculizumab and perform three time weekly dipstick testing for microscopic hematuria. Any positive test required the patient to come in for more formal testing. In 3 patients with confirmed relapse (all within 6 weeks of discontinuation of therapy), all responded quickly to re-initiation of therapy and returned to baseline creatinine. The other 7 patients had no relapses for the length of follow-up. An update revealed an average of 1 relapse per 49 months off therapy over 18 patients 21.
8.Summary
Atypical HUS is a TMA that is commonly seen in both children and adults. It can be difficult to differentiate from the other TMAs found in patients but with a good clinical history, laboratory testing, and clinical suspicion clinicians can diagnosis and quickly treat patients with atypical HUS limiting morbidity and mortality. The mainstay of treatments for atypical HUS remain similar to those for TTP including plasma exchange, steroids, and unique to atypical HUS, the use of Eculizumab. With the advent of eculizumab, patients with atypical HUS have had improvements in their renal function and have been living longer lives. More research is needed in this rare disorder to better diagnosis and treatment this potentially life threatening disease.
9.References
- Tsai HM. Untying the knot of thrombotic thrombocytopenic purpura and atypical hemolytic uremic syndrome. Am J Med 2013,126:200-9.
- Tsai HM. Pathophysiology of thrombotic thrombocytopenic purpura. Int J Hematol 2010;91:1-19.
- Ferri FD. Disseminated intravascular coagulation. Ferri’s Clinical Advisor 2016. Elsevier, 2016.
- George JN. The thrombotic thrombocytopenic purpura and hemolytic uremic syndromes: evaluation, management, and long-term outcomes experience of the Oklahoma TTP-HUS Registry, 1989-2007. Kidney Int Suppl 2009;112:S52-4.
- Coppo P, Bengoufa D, Veyradier A, et al. Severe ADAMTS13 deficiency in adult idiopathic thrombotic microangiopathies defines a subset of patients characterized by various autoimmune manifestations, lower platelet count, and mild renal involvement. Medicine 2004;83:233–244.
- Coppo P, Schwarzinger M, Buffet M, et al. Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: the French TMA reference center experience. PLoS One 2010;5:e10208.
- Balduini CL, Gugliotta L., Luppi M, et al. High versus standard dose methylprednisolone in the acute phase of idiopathic thrombotic thrombocytopenic purpura: a randomized study. Ann Hematol 2010;89:591-6.
- Hie M, Gay J, Galicier I, et al. Preemptive rituximab infusions after remission efficiently prevent relapses in acquired thrombotic thrombocytopenic purpura, Blood 2014;124:204–210.
- Te Loo DM, Monnens LA, der Velden TJ, et al. Binding and transfer of verocytoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000;95:3396-3402.
- Karpman D, Hakansson A, Perez MT, et al. Apoptosis of renal cortical cells in the hemolytic uremic syndrome: in vivo and in vitro studies. Infect. Immun. 1998;66:636-644.
- Magnus T, Rother J, Simova O, et al. The neurological syndrome in adults during the 2011 northern German E. coli serotype O104:H4 outbreak. Brain 2012;135:1850-9
- Zimmerhackl LB, Besbas N, Jungraithmayr T, et al. Epidemiology, clinical presentation, and pathophysiology of atypical and recurrent hemolytic uremic syndrome. Semin Thromb Hemost 2006;32(2):113-120.
- Atkinson JP, Liszewshi MK, Richards A, et al. Hemolytic uremic syndrome: an example of insufficient complement regulation on self-tissue. Ann N Y Acad Sci 2005;1056:144-152.
- Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 2013;8:554-562.
- Kwon T, Dragon-Durey MA, Macher MA, et al. Successful pre-transplant management of a patient with anti-factor H autoantibodies-associated haemolytic uraemic syndrome. Nephrol Dial Transplant 2008;23:2088-2090.
- Le Quintrec M, Zuber J, Noel LH, et al. Anti-factor H autoantibodies in a fifth renal transplant recipient with atypical hemolytic and uremic syndrome. Am J Transplant 2009;9:1223-1229.
- Boyer O, Balzamo E, Charbit M, et al. Pulse cyclophosphamide therapy and clinical remission in atypical hemolytic uremic syndrome with anti-complement factor h autoantibodies. Am J Kidney Dis 2010,55(5):923-927.
- Schmidtko J, Peine S, El-Housseini Y, et al. Treatment of atypical hemolytic uremic syndrome and thrombotic microangiopathies: a focus on eculizumab. Am J Kidney Dis 2013;61(2):289-299.
- Licht C, Greenbaum LA, Muus P, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney International 2015,87:1061-1073
- Ardissino G, Testa S, Possenti I, et al. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: a report of 10 cases. Am J Kidney Dis 2014;64(4):633-7.
- Ardissino G, Possenti I, Tel F, et al. Discontinuation of eculizumab treatment in atypical hemolytic uremic syndrome: an update. Am J Kidney Dis 2015;66(1):170-7.