


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
A Comparative Study of Red Blood Cell Parameters of Alpha and Beta Thalassaemia Patients Diagnosed in University Hospital in a Cheras, Malaysia
Azma RZ1, Hafiza A1, Azlin I1, Norunaluwar J1, Maizatul-Husna MA1, Joanna SKK1, Farah-Hazirah SA1, Nurul-Najiehah NM1, Najatul-Adawiyah K1, Farah-Azima AM1, Ainoon O2
2.Department of Medical Sciences II, Faculty of Medicine, Universiti Sains Islam Malaysia, Kuala Lumpur, Malaysia
Copyright : © 2018 Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Thalassaemia is the most common inherited haematological disorder in Malaysia. Red blood cell parameters analysis is the fastest and cheapest way to screen for thalassaemia trait. This study was performed to compare red blood cell parameters of alpha (α) and beta (β) thalassaemia patients diagnosed at UKM Medical Centre (UKMMC). Study was performed on 299 samples received by Haematology Laboratory, UKM Medical Centre for full blood count. Hb analysis and DNA analysis for α and β-globin gene abnormalities analysis were performed on samples with thalassaemic trait red cell indices. α and β-globin gene mutations or deletions were detected by multiplex polymerase chain reaction (PCR). Red blood cell parameters of the samples were retrieved from Intergrated laboratory management system (ILMS). Only samples with thalassaemia trait were selected for this study. The mean values for Hb, RBC, MCV and MCH were compared between α- and β- thalassaemia traits patients using student paired t-test. Out of 299, 160 (53.5%) patients were α thalassaemia trait and remaining 139 (46.5%) were β-thalassaemia trait. The most common genetic abnormalities for α-thalassaemia were αα/--SEA(48.8%) followed by αα/-α3.7 (34.7%), αα/ ααCS (4.1%), -α3.7/-α3.7 (3.5%) and αα/-α4.2 (2.9%). For β-thalassaemia, haemoglobin (Hb) E (cd26) was found to have the highest frequency (49.6%) followed by cd41/42 (18.7%), IVS1-5 (15.1%), IVS2-654 (5.7%), IVS1-1 (5.7%) and cd17 (5.04%). The overall means of Hb, RBC and MCV were significantly higher in α-thalassemia patients compared to β-thalassemia patients (p< 0.05). The red blood cell parameters for α - thalassaemia traits appeared to be higher than β-thalassaemia traits. The differences in red blood cell parameters in first line screening for thalassaemia carriers appear to be useful in the strategy for a cost-effective decision on further confirmatory tests to be done.
Thalassaemia, red blood cell parameters, mutation,Hematology
1. Introduction
Thalassaemia is one of the most common hereditary disorders of haemoglobin that occurs in most countries due to global migration, and are emerging as health problems in developing countries such as Malaysia. Thalassaemia is characterized by inefficient production of α- or β- globin chains. The defects in globin chains are due to either mutation or deletion resulting in reduced production of α- or β- globin chains which lead to a relative excess in production of the other normal globin chains. This over-production of normal globin chains causes red cells membrane instability and leads to red cells haemolysis (Bain 2006).
Screening and prevention for thalassaemia in Malaysia must be done at an extensive level which include screening for carriers, genetic counselling and facilities for prenatal diagnosis. A study by Rahimah et al. (2012) showed 7% of 310 sixteen year old secondary school students in Ampang, Kuala Lumpur were carriers for thalassaemia. While a study by Azma et al. (2012) showed 11% of 400 medical students in UKM were carriers. Another study by Ezalia et al. (2014) showed 5% of 738 healthy blood donors were presumptively diagnosed as thalassaemia carriers. Therefore, roughly 1 in 10 individuals in Malaysia is actually a carrier for thalassaemia. If these carriers are not properly counselled, it will increase the health care burden in this country as to date, there are already 6753 thalassaemia patients who require medical attention and 4782 of them are on monthly blood transfusion (National Thalassaemia Registry 2016). Most of these regularly transfused patients develop iron overload and will need regular iron chelation therapy to prevent multiorgan failure. Thus, management of patients with thalassaemia major are costly.
A successful prevention strategy includes an effective programme by health authorities and avaibility of centres with good laboratory services. Thalassaemia screening programme in Malaysia has been implemented since 2005 (Ezalia et al. 2014), however it was only targeting the antenatal mothers and cascade screening of index thalassaemia cases. Recently, in 2016 Ministry of Health, Malaysia had launched a compulsory screening programme for thalassaemia involving sixteen-year-old students in secondary schools in Malaysia.(Daily Express, Independent National Newspaper of East Malaysia 2016).
Accurate determination of carrier phenotype is essential for selection of appropriate molecular tests to determine carrier genotype. Current laboratory methods such as full blood count using fully automated systems are highly sensitive for screening for thalassaemia carriers. The measurement of red blood cell parameters such as haemoglobin (Hb), mean corpuscular haemoglobin (MCH) value, mean corpuscular volume (MCV) value, red cell distribution width (RDW) and red cell count (RCC) are useful to differentiate thalassaemia carriers from patients with underlying iron deficiency anaemia. A number of studies showed that thalassaemia patients have hypochromia (MCH <27pg) associated with eryhtrocytosis (RBC >4.75x10^12/dl) (Tripathi et al. 2015), while iron deficiency anaemia patients tend to have hypochromia with lower RBC (Soliman et al, 2015). An earlier study also showed that erythrocytes in thalassaemia trait patients were more homogenous compared to those in iron deficiency patients (Bain 2006). However, there was no study which actually specifically compared the red blood cell parameters of α to β-thalassaemia carriers. Ability to differentiate red blood cell parameters of α to β-thalassaemia carriers will help to reduce burden of government in financing for the thalassaemia investigations.
This study compared the means of red blood cell parameters of α- with β-thalassaemia patients diagnosed at UKM Medical Centre.
2. Materials And Methods
This cross-sectional and observational study was conducted at a university hospital in Cheras, Kuala Lumpur, Malaysia. Ethical approval was obtained from Institutional Review Board of UKMMC (FF-2016-221). Blood samples received by Haematology Laboratory, Department of Laboratory Diagnostics Service for thalassaemia screening from September 2015 to August 2016 were included in this study. The Hb concentration and other red blood cell parameters of the blood samples were determined using automated blood cell counter Coulter® LH 750 system (Beckman Coulter, Brea, CA, USA) and Hb analysis were performed using capillary electrophoresis (SEBIA, USA). Patients with thalassaemic red cell indices (MCH < 27pg) and HbA2 of ≥3.5% were selected for β-thalassaemia gene mutation analysis while those with MCH < 27pg but normal Hb analysis findings or with additional Hb variant were selected for -thalassaemia gene mutation analysis. Patients with concomitant iron deficiency anaemia, or known to have normochromic normocytic anaemia due to underlying chronic illness such as renal disease or autoimmune disease were excluded from the study. Patients with concomitant α and β thalassaemia were also excluded from this study. Peripheral blood sample for DNA analysis was taken after written consent was obtained.
EDTA blood samples were sent to Molecular Genetics Laboratory for DNA analysis. DNA was extracted by using QIAampR DNA Blood Mini Kit (Qiagen GmbH, Hilden, Germany) as recommended by the manufacturer and extracted DNA was stored at -30°C. The quality and concentration of the DNA was determined using NanoVue Plus spectrophotometer (GE Healthcare LtD, Buckinghamshire, UK).
DNA amplification of α-globin gene abnormalities was done using Amplification Refractory Mutation System (ARMS) method as described previously (Azma et al. 2014). Samples were subjected to multiplex gap polymerase chain reaction (PCR) amplification for the detection of common -thalassaemia gene deletions; single gene deletion -α3.7 and - α4.2; and two gene deletions --SEA, --FIL, -- MED, --(α)20.5 and --THAI (3) followed by multiplexed PCR amplification for the detection of non deletional -thalassaemia gene abnormalities: initiation codon (ATG→A-G), codon 30 (ΔGAG),codon 35 (TCC→CCC), codon 59 (GGC→GAC), codon 125 (CTG→CCG) or Hb Quong Sze and termination codon(TAA→CAA) or Hb Constant Spring). DNA amplification of β- globin gene abnormalities was performed using 5 sets of multiplex ARMS PCR and 1 set ARMS PCR to detect common β-thalassaemia point mutations: IVS 1-5 (G-C), Cd 41/42 (-TTCT), Cd 17 (A-T), Cd 26 (G-A), IVS 1-1 (G-T), Cd 8/9 (+G), -28 (A-G), Cd 71/72 (+A), IVS 1-1 (G-A), Cd 43 (G-T), Cd 16 (-C), Poly A (A-G), -88 (C-T), Initiation codon, Cd 15 (G-A), -29 (A-G), -86 (C-G), Cd 19 (A-G), Cap +1 (A-C) and IVS 2-654 (C-T) (Syahzuwan et al, 2012). All PCR products were electrophoresed on florosafe DNA stained agarose gels followed by DNA visualization under ultra-violet light illumination.
Data on the red blood cell parameters such as Hb, RBC, RDW, MCV and MCH of these α-and β-thalassaemia patients were obtained from archival data in the ILMS. SPSS was used for analysis and comparison of mean of red blood cell parameters using student paired t-test between α- and β-thalassaemia patients were done.
3. Results
A total of 299 patients with thalassaemic indices were studied. The male to female ratio was 1: 2 (89 males and 210 females). Majority of the patients were Malays (58.1%), followed by Chinese (25.4%), Indian (5%) and others (1.3%). One hundred and thirtynine (46.4%) patients were diagnosed with β- thalassaemia traits (Hb A2 > 3.5%) and 160 (53.6%) patients were α- thalassaemia traits. All patients were confirmed by DNA analysis. The range of gene abnormalities for α-thalassaemia trait include αα/--SEA (51.9%) followed by αα/-α3.7 (36.9%), αα/ ααCS (4.4%), -α3.7/-α3.7 (3.8%) and αα/-α4.2 (3.1%) (Table1).
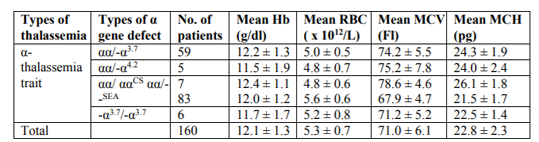
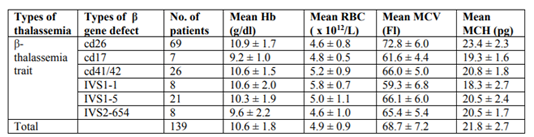
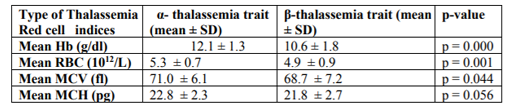
For the β-thalassaemia trait group, the range of gene abnormalities include cd26 (G-A) (49.6%) followed by cd41/42 (-TTCT) (18.7%), IVS1-5 (G-C) (15.1%), IVS2-654 (C-T) (5.7%), IVS1-1(G-T) (5.7%) and cd17 (A-T) (5.04%) (Table 2). Results of the red blood cell parameter analysis showed the overall means of Hb, RBC, MCV and MCH for β-thalassaemia traits were much lower compared to α- thalassaemia traits (Tables 1 and 2). Statistical analysis showed significant differences in the means of Hb, RBC and MCV between α- and β-thalassaemia patients (p < 0.05). There was no significant difference in the means of MCH between α- and β-thalassaemia patients (p > 0.05) (Table 3).
4. Discussion
Retrospective screening has been performed in Malaysian populations, which includes pregnant women during antenatal check-up and relatives of thalassaemia patients. This method of screening is useful for populations with a very low frequency of thalassaemia. However, this method is no longer applicable since Malaysian population is now known to have a high incidence of thalassaemia carriers (Azma et al. 2012, Ezalia et al. 2014) and the number of thalassaemia patients who required regular blood transfusion is reaching 5000 patients (National Thal Registry 2016). Recent effort by Ministry of Health, Malaysia in implementing thalassaemia screening programme for sixteen-year-old secondary school students will definitely improve future health burden. All patients who are confirmed to have thalassaemia receive genetic counselling before they plan to get married and have family.
Thalassaemia and iron deficiency anaemia are recognized as the most important causes of hypochromic microcytic red blood cell parameters. Numerous studies have shown that thalassaemia carriers were easily differentiated from iron deficiency anaemia based on red blood cell parameters. In a study by Soliman et al. (2014), iron deficiency anaemia group showed a highly significant reduction in the mean of Hb level when compared with the normal donor group, whereas the mean of Hb in the β thalassaemia trait group showed no significant difference compared to the normal donor group. This result proved that the β thalassaemia trait group had a higher Hb concentration and a lesser degree of anaemia compared to the iron deficiency anaemia group. Another useful parameter in which a significance difference was seen between thalassaemia carrier and IDA was RDW. IDA patients tend to have higher value of RDW compared to thalassaemia carrier. This is because; red cells in thalassemia carriers were more homogeneous than those of IDA patients (Matos et al, 2015). However, there was no study comparing red blood cell parameters between α and β-thalassaemia carriers. Knowledge of the red blood cell parameters of thalassaemia carriers will facilitate the decision on appropriate additional tests for confirmation of diagnosis, hence saving laboratory costs.
In this study, statistical analysis showed significant differences in the means of Hb, RBC and MCV between and -thalassaemia carriers (p < 0.05). The mean of MCH in β-thalassaemia was slightly lower compared to -thalassaemia even though it was statistically not significant. These markedly differences of red cell indices are due to excess of and gamma () globin chains in -thalassaemia trait that form partially soluble but ineffective hemoglobin homotetramers (-chain tetramers). These -chain tetramers, which are also referred as Hb H will precipitate extensively when they are exposed to oxidant stress and lead to lysis of circulating RBC. While in -thalassaemia trait, free -chains are unstable and do not form tetramers. They are easily precipitated adjacent to the RBC membrane. These precipitated -chains form inclusions in both RBC precursors and circulating RBCs which cause premature haemolysis of cells in bone marrow as well as in peripheral circulation (Bain 2006).
Hb analysis has been useful for decades and most of -thalassaemia trait patients have successfully been detected by this method. However, the test is not useful in the detection of -thalassaemia carrier and most of these patients have been missed for years. Even though Hb Constant Spring may appear in Hb analysis, quite a number of heterozygotes patients have been missed using High Performance Liquid Chromatography (Raja Zahratul et al. 2016). Detection of Haemoglobin H (HbH) inclusion bodies was previously to diagnose -thalassaemia. However, the sensitivity of this test was often unsatisfactory and a study by Wang et al. (2000) showed none of single or two -gene deletions thalassaemia patients showed positive results in this test. Only three and four gene deletion forms of -thalassaemia will have positive results with HbH inclusion. Understanding their red blood cell parameters characteristic may be useful and could potentially reduce the cost by eliminating the need of running multiple tests such as Hb analysis and HbH inclusion. DNA analysis for -thalassaemia can directly be performed for confirmation.
Molecular analysis is highly accurate but laborious and may require multiple steps of analysis. As seen in this study, in -thalassaemia, IVS 1-5 (G-C), Cd 41/42 (-TTCT) and Cd 26 (G-A) were among the commonest mutations seen in Malaysian population (George et al. 2010) and the multiplex PCR kit was designed to detect these mutations in a single analysis. However, the less common mutations such as IVS2-654 (C-T), IVS1-1(G-T) and Cd 17 (A-T), required additional runs, since primers for detection of these mutations were designed in a different kit. Similar situation was seen in -thalassaemia patients, αα/--SEA and αα/-α3.7 could easily be in single run but for less common alpha gene abnormalities such as Hb Constant Spring, additional analysis was necessary.
5. Conclusions
A practical approach using full blood count is cheaper and faster especially when extensive screening for Malaysian population is undertaken. This study has shown that, α and β-thalassaemia carriers can be differentiated using red blood cell parameters. The accurate determination of the carrier phenotype is essential for selection of appropriate confirmatory molecular tests.
References
- Azma, R.Z., Ainoon, O., Azlin, I., Hamenuddin, H., Hadi, N.A., Tatt, W.K., Syazana, I.N., Asmaliza, A.M., Das, S., Hamidah, N.H. 2012. Prevalence of iron deficiency anaemia and thalassaemia trait among undergraduate medical students. Clin Ter163(4): 287- 291.
- Azma, R.Z., Ainoon, O., Hafiza, A., Azlin, I., Noor Farisah, A.R., Nor Hidayati, S., Noor Hamidah, H. 2014. Molecular characteristic of alpha thalassaemia among patients diagnosed in UKM Medical Centre. Malays J Pathol37 (1): 27-32.
- Bain, B.J. 2006. The α, β, δ and γ thalassaemias and relatedconditions. In Haemoglobinopathy Diagnosis. 2nd edition. Oxford: Blackwell Publishing; 63-127.
- Daily Express, Independent National Newspaper of East Malaysia. 2016. Thumbs up for free thalassaemia screening. http://www.dailyexpress.com.my/news.cfm?Ne wsID=111718 [31 July 2016]
- Ezalia, E., Irmi Elfina, R., Elizabeth, G., Wan Hayati, M.Y., Norhanim, A., Wahidah, A., Chin, Y.M., Rahimah, A., Nor Asiah, M., Masita, A., Rohaidah, H., Zubaidah, Z. 2014. Thalassaemia Screening among Healthy Blood Donors in Hospital Tengku Ampuan Rahimah, Klang Med & Health9 (1): 44-52.
- George, E., Mary Ann, T.J.2010. Genotype-phenotype diversity of beta thalassaemia in Malaysia: Treatment options and emerging therapies. Med J Malaysia. 65(4); 256 – 259.
- Matos, J.F., Borges, K.B.G., Fernandes, A.P.S.M., Faria, J.R., Carvalho, M.d.G. RDW as differential parameter between microcytic anemias in “pure” and concomitant forms. 2015. J Bras Patol Med Lab. 51 (1); 22 – 27.
- National Thalassaemia Registry. 2016. My Talasemia MalaysiaThalassaemia Vortal, Ministry of Health Malaysia. (Unpublished data).
- Rahimah, A.N., Nisha, S., Safiah, B., Roshida, H., Punithawathy, Y., Nurul, H., Syahzuwan, H., Zubaidah, Z. 2012. Distribution of alpha thalassaemia in 16-year-old Malaysian students in Penang, Melaka and Sabah. Med J Malaysia67 (6): 565-570.
- Raja Zahratul, A., Khamisah, M.G., Suria, A.A., Hafiza, A., Azlin, I., Noor Farisah, R., Nor Hidayati, S., Malisa, M.Y., Zarina, A.L., Hamidah, A., Endom, I., Danny, K.X.R., Ainoon, O.2016. Detection of homozygous Haemoglobin Constant Spring by capillary electrophoresis method.ARC Journal of Haematology 1 (1): 28-32.
- Hassan, S., Ahmad, R., Zakaria, Z., Zulkafli, Z. & Abdullah, W. Z. 2013. Detection of β-globin Gene Mutations Among β-thalassaemia Carriers and Patients in Malaysia: Application of Multiplex Amplification Refractory Mutation System–Polymerase Chain Reaction. Malays J Med Sci 20(1): 13 – 20.
- Soliman, A.R., Kamal, G., Elsalakawy, A., Mohamed, T.H. 2014. Blood indices to differentiate between Beta-Thalassaemia trait and iron deficiency anaemia in adult healthy Egyptian blood donors. Egypt J Haematol39: 91-92.
- Tripathi, N., Soni, J.P, Sharma, P.K, Verma, M. 2015. Role of Haemogram Parameters and RBC Indices in Screening and Diagnosis of Beta-Thalassaemia Trait in Microcytic, Hypochromic Indian Children. International Journal of Hematological Disorders 2(2): 43-46.
- Wang, C., Beganyl, L., Fernandes, B.J. 2000. Measurements of red cell parameters in alpha thalassaemia trait: Correlation with the genotype. Lab Hematol6: 163-166.