


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Clinical Characteristics of Patients Diagnosed with Delta Granule Platelet Storage Pool Deficiency (Δ-PSPD) at the Detroit Medical Center (DMC), Detroit MI
Indryas L.Woldie1,Robert Guo2,Rosanne Ososki3,Greg Dyson4,Shahid Mohamad5,Kunil K.Raval6,Ali Gabali7
2.University of Western ON, Schulich School of Medicine, USA .
3.Detroit Medical Center, Detroit MI, USA.
Copyright : © 2017 . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Delta granule platelet storage pool deficiency (δ-PSPD) is a form of bleeding disorder that results from decrease in the average number of delta granules in platelets. The objective of this study was to describe clinical characteristics of patients with δ-PSPD. Retrospective data from CDC surveillance data set was collected for 56 patients diagnosed with δ-PSPD from Jan. 2005-May 2012.The commonest bleeding complaint was excessive bruising. There was no significant association between severity of platelet dense granulecount and bleeding phenotype. For the 18 patients for which light transmission aggregometry (LTA) data was available, there was no correlation between LTA findings and platelet dense granule count. In addition, significant proportion of patients also had arterial or venous thrombosis. Additional studies are desperately needed to define and standardize the diagnosis and management of δ-PSPD.
Clinical Characteristics,Patients Diagnosed,Delta Granule Platelet Storage,Detroit Medical Center (DMC), Detroit MI,Hematology
1. Introduction
Platelets are cells that are essential for maintaining hemostasis. They contain numerous substances that mediate both clotting and thrombolysis. These substances are stored principally in two types of organelles, alpha and delta (dense) granules [20]. Storage pool deficiency (SPD) is a heterogeneous bleeding disorder related to a decreased number of these specific platelet organelles resulting in platelet dysfunction. Patients having SPD often present with a history of easy bruising, frequent epistaxis, and at times with positive family history [21].
Delta granule platelet storage pool deficiency (δ-PSPD) is a form of bleeding disorder that results from decrease in the average number of delta granules in platelets. Both acquired and congenital forms of δ-PSPD have been described [1-4]. However; there is no uniform consensus on its clinical presentation, correlation with available platelet function studies and optimal management.
Currently the diagnosis of δ-PSPD is made by Electron Microscopy and clinical symptoms [1]. Both PFA and platelet agregometry were not found to be reliable tests to predict bleeding in patients with δ-PSPD [6,8] Recent case series on 16 patients with δ-PSPD showed moderate abnormalities with variable profiles in platelet aggregation, while all the individuals had almost complete absence of adenosine triphosphate release. In this series mepacrine capture, CD63 expression, and study of dense granules by electron microscopy enabled to distinguish different subtypes of δ-SPD with quantitative or qualitative defect [11]. However there is lack of a standardized diagnostic algorithm and the literature so far is based mainly on case series of few patients at individual centers.
2. Objectives
The objective of the study is to describe clinical characteristics of patients with delta granule platelet storage pool deficiency with respect to bleeding phenotype (site, severity), clotting phenotype (arterial or venous), treatment used, if any, and treatment outcome. In addition, pattern of platelet aggregation studies will be explored whenever available.
3. Design And Methods
Retrospective record review was made from ATHN/CDC surveillance data base for patients diagnosed with δ-PSPD at the DMC from 2005-2012. The ATHN data set is a surveillance data set with no identifiable patient information and was already approved as exempt by the local Institutional Review Board. Patients with concomitant bleeding disorder were excluded. A total of 56 patients with average platelet dense granule count of < 3.75, diagnosed with δ-PSPD were identified. Data on clinical presentation particularly bleeding phenotype (site and severity), history of arterial or venous thrombosis, treatment used (if any) and its effectiveness were collected. Descriptive statistics were made and associations were looked into whenever appropriate.
4. Light Transmission Aggregometry
The protocol was adapted from a standard operating procedure of the Detroit Medical Center University Laboratories – Core Coagulation for clinical diagnostics. ADP and Epinephrine (Helena Laboratories, Beaumont, TX) were used to induce platelet aggregation. Light transmission was measured on a BIO/DATA PAP-4 aggregometer (BIO/DATA, Horsham, PA) coupled with a Chrono-log Aggregometer Model 700 sample chamber. Light transmission was measured at a fixed wavelength in the infrared spectrum.
5. Sample Preparation
Blood from patients who had not taken aspirin containing products for 10-14 days was collected in 3.2% sodium citrate buffer and maintained at room temperature and gently rocked for 6 minutes. The blood was then spun at 100 x g for 10 minutes to produce platelet rich plasma (PRP) supernatant. Approximately ¾ of the PRP supernatant was collected and the remaining ¼ PRP supernatant and solids were spun again at 1400 x g for 20 minutes to produce platelet poor plasma (PPP) supernatant. The PRP and PPP were < 250x109 and < 10x109platelets/L, respectively.
6. Assay Protocol
Testing was performed at 37oC under stirred conditions in either plastic or siliconized glass cuvettes. 100% and 0% transmission were defined by the light transmitted through PPP and PRP, respectively. Aggregation was measured by adding ADP or Epinephrine to PRP to a final concentration of 100 nM each. The % light transmission was then recorded for 7 minutes. Controls to test for spontaneous aggregation were run in parallel. Each sample was run in triplicate.
7. Data Analysis
Data was collected as % transmission vs. time. All samples reached a stable % transmission/ time (slope = 0) within 7 minutes. The final value reported, % maximal aggregation, was calculated as the amount of light transmitted through the sample at equilibrium relative to PPP, which was set at 100%.
Light transmission aggregometry (LTA) results for ADP and Epinephrine were available for a subset of 18 patients on which exploratory analysis was made. LTA results for ADP and Epinephrine were interpreted using the following cut off points: hypo-responsiveness (decreased) for aggregation < 39% and hyper-responsiveness (increased) for aggregation >79%. Aggregation between 39%-79% was defined as normal.
Statistical analyses were performed using SPSS v20 (IBM; Armonk, NY). δ-PSPD was diagnosed using electron microscopy as described by White J.G. and reported as the average number of dense granules per platelet after reviewing 100 platelets[5].δ-PSPD was further classified as mild (PDG between 3-3.75), moderate (PDG between 2-2.99) and severe (PDG between 0-1.99). Patients were usually evaluated because of personal history of bleeding or bruising. However, few patients were also seen because of family history of δ-PSPD.
8. Results
A total of 56 patients with δ-PSPD were identified. Of these, 93% were females. Majority (64%) were Caucasians and 36% were African Americans. With respect to severity, majority (59%) had moderate δ-PSPD (PDG 2-2.99) followed by 29% who had mild δ-PSPD (PDG 3-3.75) and the rest 12% had severe δ-PSPD (PDG 0-1.99).
Table1: Patient Characteristics and Demographics (N = 56)
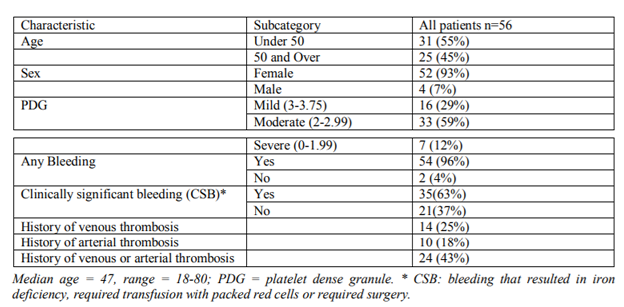
Almost all patients (96%) had some form of bleeding history which was the reason for their clinic visit. The other 4% came because of family history of bleeding disorder but without personal history of bleeding. [Table 1]
Excluding those patients with incomplete information on the various types of bleeding and those with no previous surgical challenge, the commonest bleeding complaint was excessive bruising (94%), followed by menorrhagia (90%), gum bleeding (63), epistaxis (53%), post-operative hemorrhage (51%), dental procedures (47%) and post-partum hemorrhage (44%).
Clinically significant bleeding (CSB), defined as bleeding that resulted in iron deficiency, required blood transfusion and/or surgical intervention, was reported in 63% of patients.
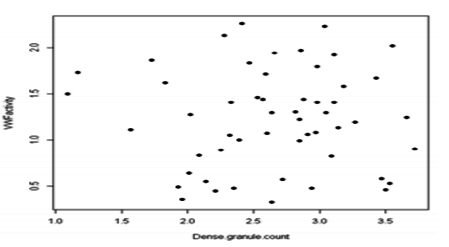
There was no significant association between severity of platelet dense granulecount and bleeding phenotype. Although all patients in this study have normal VWF activity, there was no correlation between dense granule count and VWF activity levels (Figure 1).
Figure1. Dot plot showing no correlation between VWF (Von Wilbrand’s Factor) activity and dense granule count
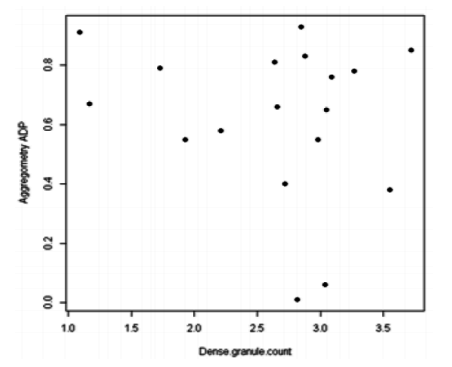
On the 18 patients for which LTA data was available; 6, 7 and 5 had normal (39-79%), increased (>79%) and decreased (< 39%) aggregometry for EPI respectively. Whereas 10, 6 and 2 had normal, increased and decreased aggregometry for ADP respectively. There was no correlation between LTA findings (either Epinephrine or ADP) and platelet dense granule count (Figs. 2 and 3).
Figure2. Dot plot showing no correlation between platelet aggregometry with EPI (Epinephrine) and dense granule count
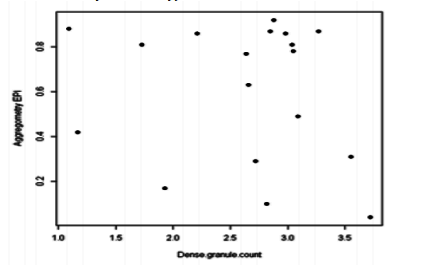
Figure3. Dot plot showing no correlation between platelet aggregometry with ADP (adenosine di-phosphate) and dense granule count
9. Discussion
The study showed significant female predominance (>90%) for δ-PSPD which was also reported on previous studies [11]. This is probably due to menorrhagia in women leading to more evaluation and/or referral. However, the presence or absence of additional biologic explanation remains to be investigated. The fact that more than half (63%) of patients reported clinically significant bleeding suggests δ-PSPD is probably a more significant bleeding disorder that requires more attention. Our study also showed a lack of association between LTA and platelet dense granule count as previously described [6,7]. Similarly, studies have shown that the platelet function assay (PFA 100) does not predict delta granule platelet storage pool deficiency [8]. This is an area that warrants further research to identify a simple, reliable and reproducible platelet function test that better correlates with patients’ clinical phenotype. Secretion aggregometry, which measures the degree of ATP release from platelet dense granules was found to show considerable variability even among healthy subjects in one study[9]. On the contrary, studies by Pai et.al and Selle et.al. suggested that ATP release assays are useful regardless of conventional platelet aggregation findings [10,11]. Another test that is advocated by several investigators is mepacrine based flow cytometry [2,12-14]. Gordon et.al. Described simple mepacrine based flow cytometry method for the detection of acquired and congenital δ-PSPD. Conventional aggregometry was normal in those patients and flow cytometry studies showed good correlation with electron microscopy findings [12]. An interesting study recently published by Cai et.al. combined flow cytometric mepacrine uptake/release with CD 63 assay and was able to differentiate between delta granule storage (δ-PSPD) and release disorders [15]. Some authors have also described the use of thromboelastogram in the management of patients with δ-PSPD [16]. Radioactive serotonin uptake and release assay is another test mainly used for research purposes but not clinical practice [17]. Use of next generation sequencing to identify mutations in inherited platelet function defect was also explored in one study and showed tremendous heterogeneity/ genetic complexity [18]. In summary mepacrine based flow cytometry appears to be attractive and relatively simple test that needs further standardization to be used as a one of diagnostic tools for δ-PSPD.
Another observation of our study is the lack of correlation between severity of bleeding and severity of platelet dense granule deficiency. This is of particular concern as it will make treatment plan difficult, particularly for those patients who have no previous surgical challenge. Literature has suggested that delta granules may not be absolutely essential for platelet function based on the observation that some patients do not have bleeding despite absence of dense granules [19]. This suggests that additional factors may contribute to a bleeding phenotype in patients with δ-PSPD and remains to be investigated.
In contrast to what was expected, close to half (43%) of the patients with δ-PSPD were found to have venous or arterial thrombosis. This creates a diagnostic as well as management dilemma, as those patients are now in need of anticoagulation or anti platelet agents, both of which are counterintuitive and possibly harmful in patients with a potential bleeding disorder. Unfortunately, our data was insufficient to find out if the arterial or venous thrombosis diagnosis preceded the diagnosis of δ-PSPD or vice versa.
Currently there is no consensus on management of patients with δ-PSPD, particularly with respect to pre-surgical planning. Some centers use bleeding phenotype as a guide to gauge treatment which could vary from recommending platelet transfusion with or without anti-fibrinolytics, DDAVP, or even no intervention. This lack of uniformity in treating δ-PSPD shows the need for more research in the hope of getting more understanding that will help develop consensus guideline. Evidence based practice will help avoid unnecessary exposure to platelets, unnecessary cost to the health care system as well as dangers of excessive bleeding in patients with δ-PSPD who may need to be pre treated.
References
- Kakali Ghoshal, Maitree Bhattacharyya. Overview of Platelet Physiology: Its Hemostatic and Nonhemostatic Role in Disease and Pathogenesis. The Scientific World Journal, Volume 2014 (2014)
- Shirley M. Abraham, Koh Boayue , Ibraahim Ahmed. Platelet Delta Storage Pool Deficiency In Children: A case Series.
- Nayak L, Schmaier AH. A platelet acquired storage pool disorder associated with tamoxifen therapy. Case Rep Hematol. 2012;2012: 948351.
- Billio A, Moeseneder C, Donazzan G, Triani A, Pescosta N, Coser P. Hermansky-Pudlak syndrome: clinical presentation and confirmation of the value of the mepacrine-based cytofluorimetry test in the diagnosis of delta granule deficiency. Haematologica. 2001; 86(2):220.
- Gerrard JM, McNicol A. Platelet storage pool deficiency, leukemia, and myelodysplastic syndromes. Leuk Lymphoma. 1992;8(4-5):277-81.
- Mouly S, Youssefian T, Souni F, Cramer E, Lefrere F, Varet B, et al. Acquired delta-storage pool deficiency associated with idiopathic myelofibrosis. Leuk Lymphoma. 2000;37(5-6):623-7.
- White JG. Use of the electron microscope for diagnosis of platelet disorders. Seminars in thrombosis and hemostasis. 1998;24(2):163-8.
- Woods GM, Kudron EL, Davis K, Stanek J, Kerlin BA, O'Brien SH. Light Transmission Aggregometry Does Not Correlate With the Severity of delta-Granule Platelet Storage Pool Deficiency. J Pediatr Hematol Oncol. 2016; 38(7):525-8.
- Nieuwenhuis HK, Akkerman JW, Sixma JJ. Patients with a prolonged bleeding time and normal aggregation tests may have storage pool deficiency: studies on one hundred six patients. Blood. 1987; 70(3):620-3.
- Sladky JL, Klima J, Grooms L, Kerlin BA, O'Brien SH. The PFA-100 (R) does not predict delta-granule platelet storage pool deficiencies. Haemophilia. 2012;18(4):626-9.
- Badin MS, Graf L, Iyer JK, Moffat KA, Seecharan JL, Hayward CP. Variability in platelet dense granule adenosine triphosphate release findings amongst patients tested multiple times as part of an assessment for a bleeding disorder. International journal of laboratory hematology. 2016.
- Pai M, Wang G, Moffat KA, Liu Y, Seecharan J, Webert K, et al. Diagnostic usefulness of a lumi-aggregometer adenosine triphosphate release assay for the assessment of platelet function disorders. American journal of clinical pathology. 2011; 136(3):350-8.
- Selle F, James C, Tuffigo M, Pillois X, Viallard JF, Alessi MC, et al. Clinical and Laboratory Findings in Patients with delta-Storage Pool Disease: A Case Series. Seminars in thrombosis and hemostasis. 2016.
- Gordon N, Thom J, Cole C, Baker R. Rapid detection of hereditary and acquired platelet storage pool deficiency by flow cytometry. British journal of haematology. 1995; 89(1):117-23.
- Ramstrom AS, Fagerberg IH, Lindahl TL. A flow cytometric assay for the study of dense granule storage and release in human platelets. Platelets. 1999;10(2-3):153-8.
- Wall JE, Buijs-Wilts M, Arnold JT, Wang W, White MM, Jennings LK, et al. A flow cytometric assay using mepacrine for study of uptake and release of platelet dense granule contents. British journal of haematology. 1995;89(2):380-5.
- Cai H, Mullier F, Frotscher B, Briquel ME, Toussaint M, Massin F, et al. Usefulness of Flow Cytometric Mepacrine Uptake/Release Combined with CD63 Assay in Diagnosis of Patients with Suspected Platelet Dense Granule Disorder. Seminars in thrombosis and hemostasis. 2016; 42(3):282-91.
- Campbell J, Yentis S. The use of thromboelastography for the peripartum management of a patient with platelet storage pool disorder. Int J Obstet Anesth. 2011; 20(4):360; author reply 1.
- Mumford AD, Frelinger AL, 3rd, Gachet C, Gresele P, Noris P, Harrison P, et al. A review of platelet secretion assays for the diagnosis of inherited platelet secretion disorders. Thromb Haemost. 2015;114(1):14-25.
- Leo VC, Morgan NV, Bem D, Jones ML, Lowe GC, Lordkipanidze M, et al. Use of next- generation sequencing and candidate gene analysis to identify underlying defects in patients with inherited platelet function disorders. Journal of thrombosis and haemostasis : JTH. 2015; z13(4):643-50.
- White JG. Platelet granule disorders. Critical reviews in oncology/hematology. 1986;4(4): 337-77